|
ABOUT
The problem of DNA sequence classification is central to several application domains in molecular
biology, genomics, metagenomics and genetics. Although several software tools have been developed for this problem,
it is still computationally challenging due to the
size of datasets generated by modern sequencing instruments and the growing size of reference
sequence databases.
We present CLARK, a method
based on a supervised sequence classification using discriminative k-mers.
Considering two distinct specific classification
problems (see the article for details), namely (1) the taxonomic classification of metagenomic reads to known bacterial genomes, and
(2) the assignment of BAC clones and transcript to chromosome arms/centromeres
(in the absence of a finished assembly for the reference genome), CLARK outperforms
in classification speed and precision the best state-of-the-art methods.
Three classifiers from the CLARK framework are provided:
-
CLARK (default): created for powerful workstation,
it can require a significant amount of RAM to run with large database (e.g., all bacterial genomes from
NCBI/RefSeq). This classifier is the standard in the CLARK tool series.
It builds discriminative k-mers from all k-mers in the targets, queries k-mers with exact matching,
and, in its fastest mode, classifies 1 million short reads in few seconds...;
-
CLARK-l : created for workstations with limited memory
(i.e., "l" for light), this software tool provides precise classification on small metagenomes.
Indeed, for metagenomics analysis, CLARK-l works with a sparse or ''light'' database
(up to 4 GB of RAM) while still performing ultra accurate and fast results.
This classifier builds discriminative k-mers from non-overlapping and distant k-mers in the targets and queries k-mers with exact matching;
-
CLARK-S: created for powerful workstations and exploiting spaced
k-mers (i.e., "S" for spaced), this classifier requires a higher
RAM usage than CLARK or CLARK-l, but it does offer a higher sensitivity than CLARK
at the species level (see the peer-reviewed publication in Bioinformatics).
CLARK-S completes the series of classifiers from the CLARK framework.
Other applications of CLARK are, for example, the detection of contaminants, the identification of chimerism and vector contamination in sequenced BACs (cf. "Overview" tab).
|
PERFORMANCE
First, in the context of BACs assignments, CLARK shows a high discriminative power when assigning sequences
to chromosome arms and, for the first time, centromeres labels to 15K barley BACs and 51K barley unigenes.
The analysis of the confidence score of these assignments shows an average score of 0.924 which
indicates that the number of hits for the top target (which will receive the assignment) is about 12 times
higher than the second.
Second, in the context of metagenomic classification (using the bacterial/archaeal genomes of NCBI/RefSeq),
extensive experimental results show that CLARK's classification precision and sensitivity, at the genus/species level,
are better or equivalent than best state-of-the-art tools, and it is significantly faster than any published methods.
CLARK, in its fastest mode,
can classify short sequences of a simulated metagenome ("HiSeq", cf. "Simulated Datasets" below),
at the speed of 32 million reads per minute (single-threaded task),
which is at least 5 times faster than Kraken
(which is itself several order of magnitude faster than Megablast or other standard tools).
While faster, CLARK also achieves ultra-high precision and
requires less memory space in disk than Kraken or NBC (see the following table).
Finally, the analysis of the confidence score of these assignments shows an average confidence score of 0.998.
This implies that the number of hits for the top target is in average about 585 times
higher than the second.
From the following table, we can compare NBC and Kraken against CLARK
based on their accuracy speed (i.e., the number of correct reads produced per unit of time, or also speed x sensitivity).
Observe that CLARK outperforms the other tools, and can produce 2.4 million of correct reads per minute.
|
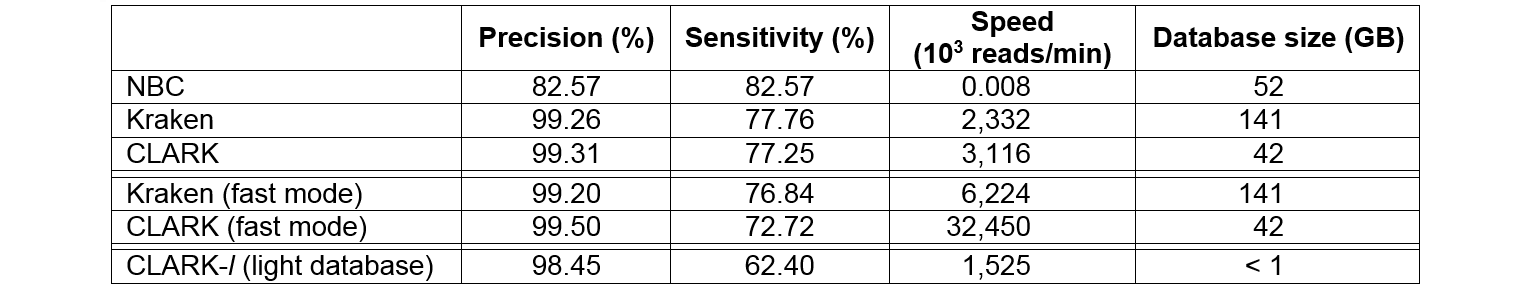 |
Classification performance (precision and
sensitivity at the genus level, computational speed, and memory space required
in disk to store the database) of best state-of-the-art metagenomics tools,
Naive Bayesian Classifier or NBC(v1.1, N=15) and Kraken (v0.10.4-beta, k=31) against
CLARK (v1.0, k=31). The results are for HiSeq, a simulated sample
of 10,000 reads (average reads length of 92 bp) and in single-threaded mode.
All experiments were run on a Dell PowerEdge T710 server (dual
Intel Xeon X5660 2.8 Ghz, 12 cores, 192 GB of RAM), except for CLARK-l, which
was run on a Mac OS X, Version 10.9.5 (2.53 GHz Intel Core 2 Duo, 4 GB of RAM).
Update:
We have rerun these evaluations with the releases v0.10-5 and v0.10-6 and
we did not find any discrepancies with the version initially used.
|
SIMULATED DATASETS
(A) In the BMC Genomics paper (2015), we have tested CLARK against four simulated metagenomes. The three datasets "HiSeq", "MiSeq" and "simBA-5"
from the Kraken project were used, as well as the set of simulated reads, "simHC", from the FAMeS project
(Fidelity of Analysis of Metagenomic Samples),
which simulates high complexity communities lacking dominant populations.
The simHC dataset contains 113 subsets
of long Sanger reads from various known microbial genomes. We have chosen arbitrarily 20 subsets
corresponding to 20 distinct genomes (see the following table), and extracted the first 500 reads to build a set of 10,000 reads.
We call this resulting set of reads "simHC.20.500". We list below the 20 genomes
we used, identified by their specific ID (Integrated Microbial Genomes ID) and accessible in the Joint
Genome Institute (JGI) database. To download this dataset, go to the "FAQ" tab (Q12).
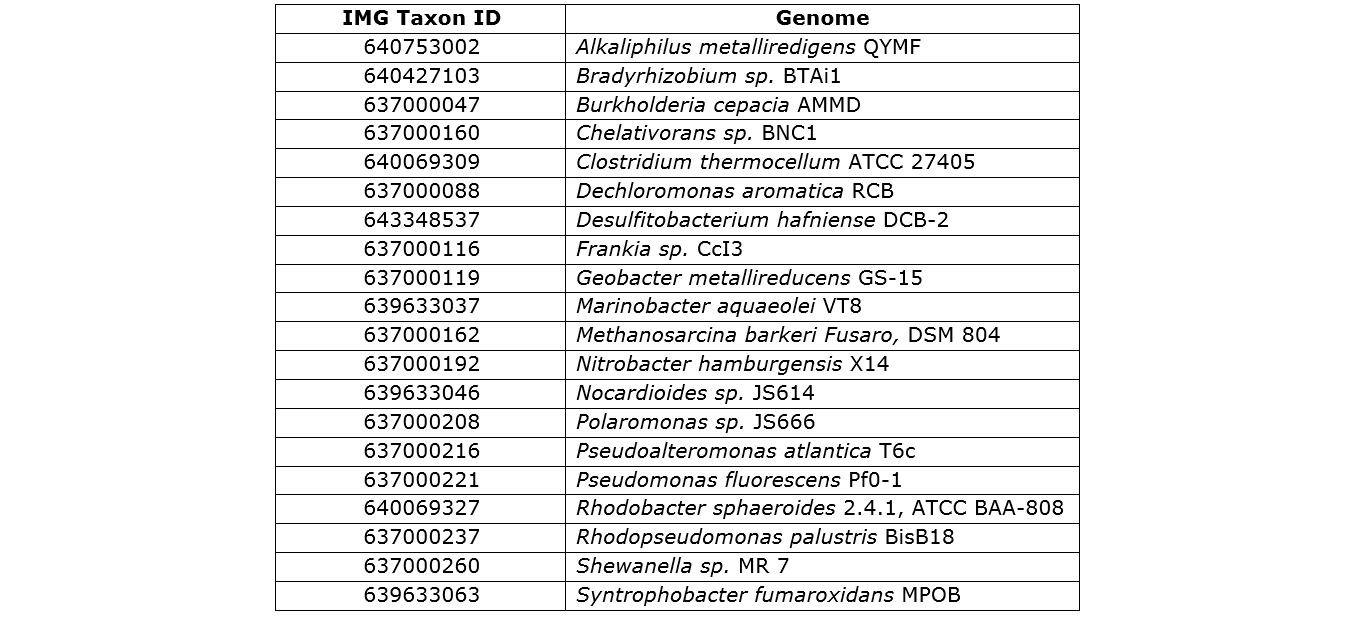
Component genomes from the JGI database used in the "simHC.20.500" dataset.
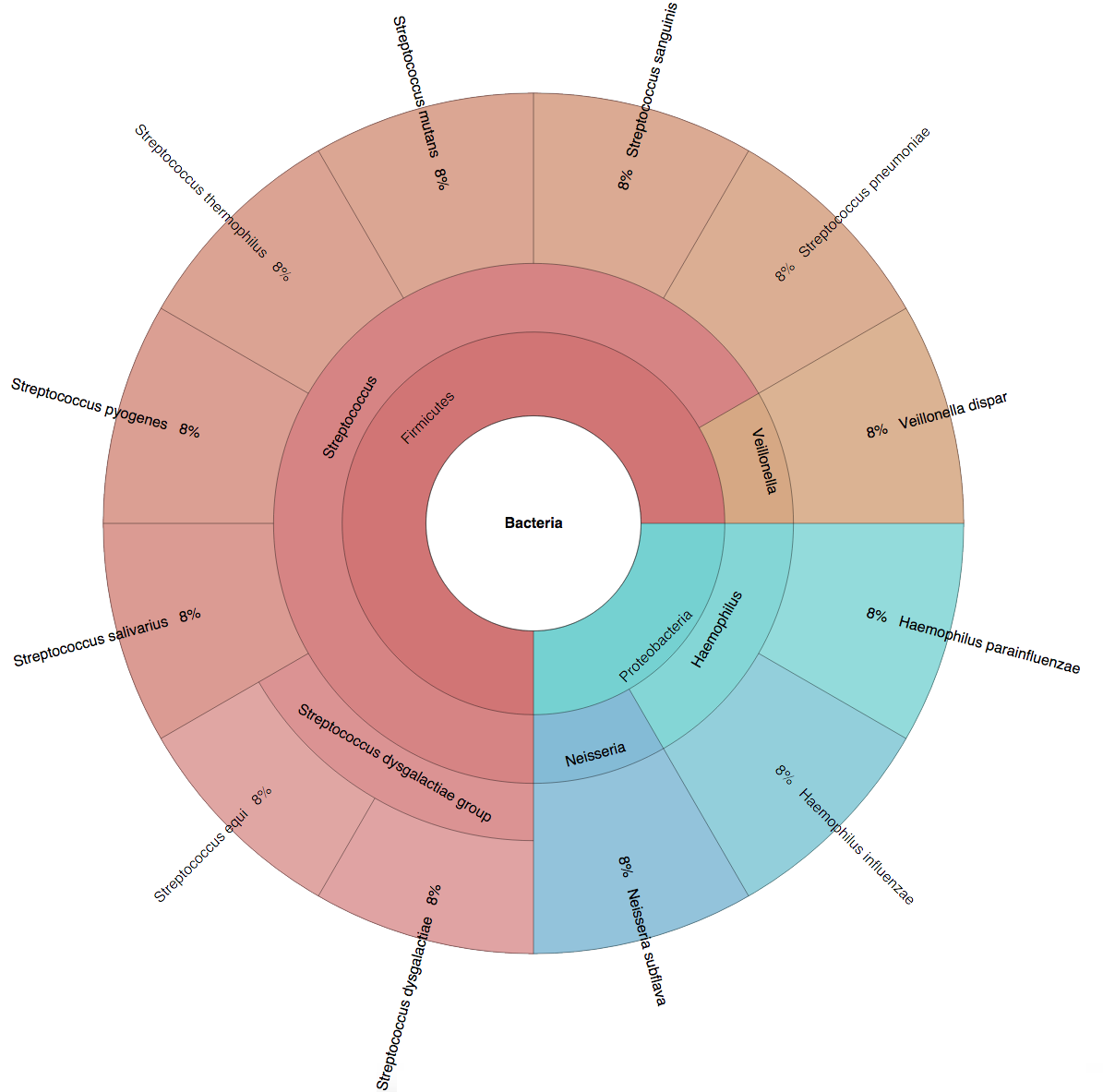
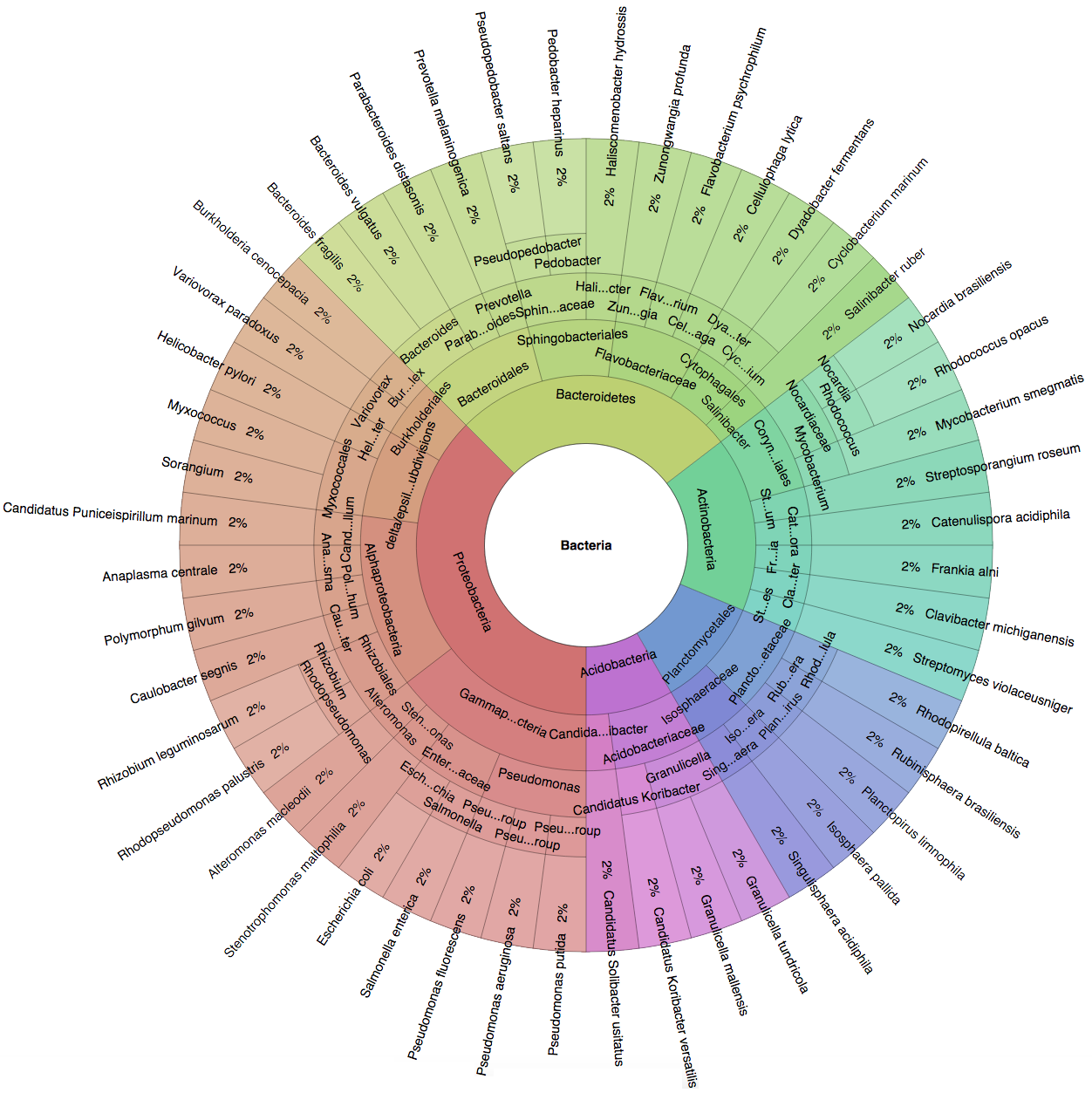
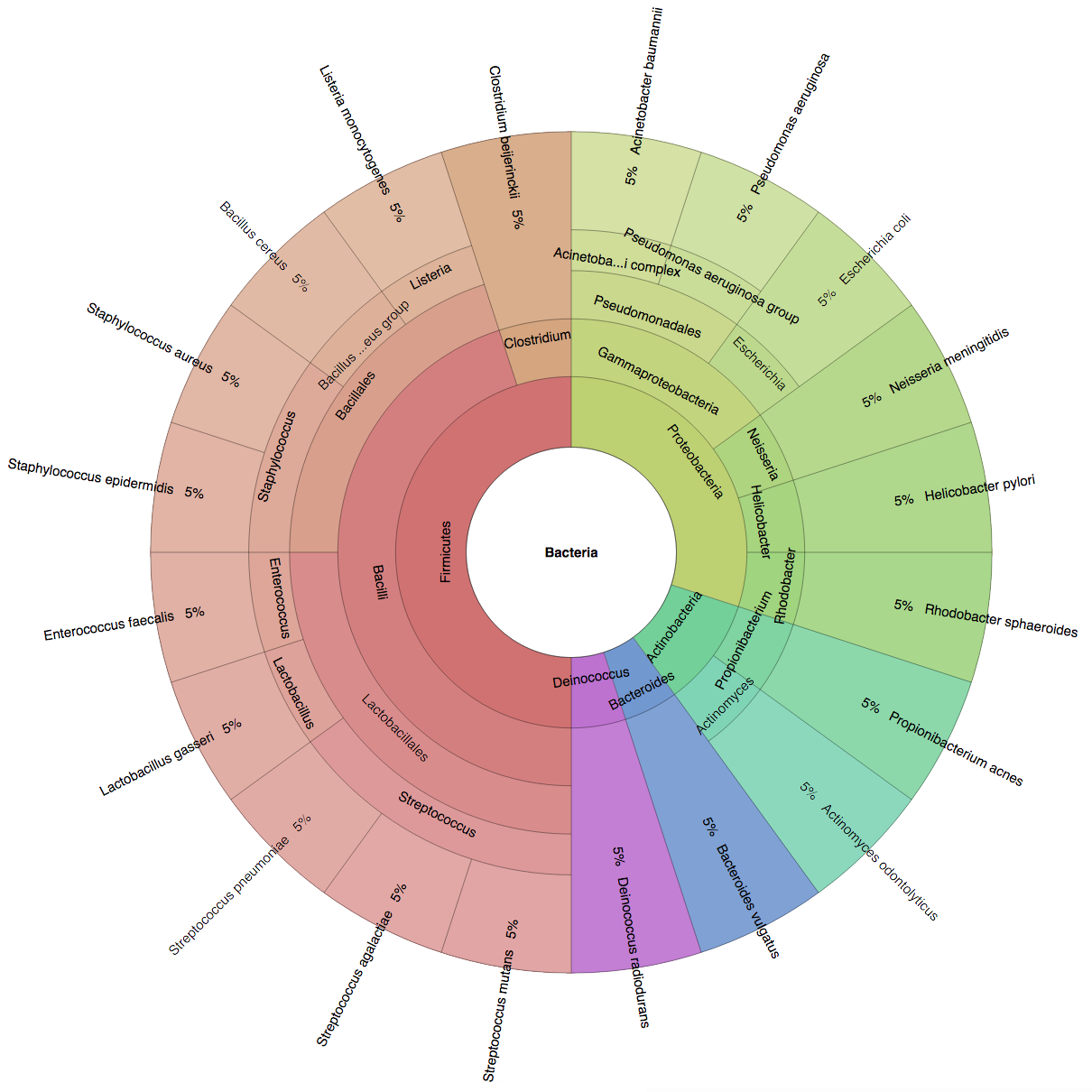
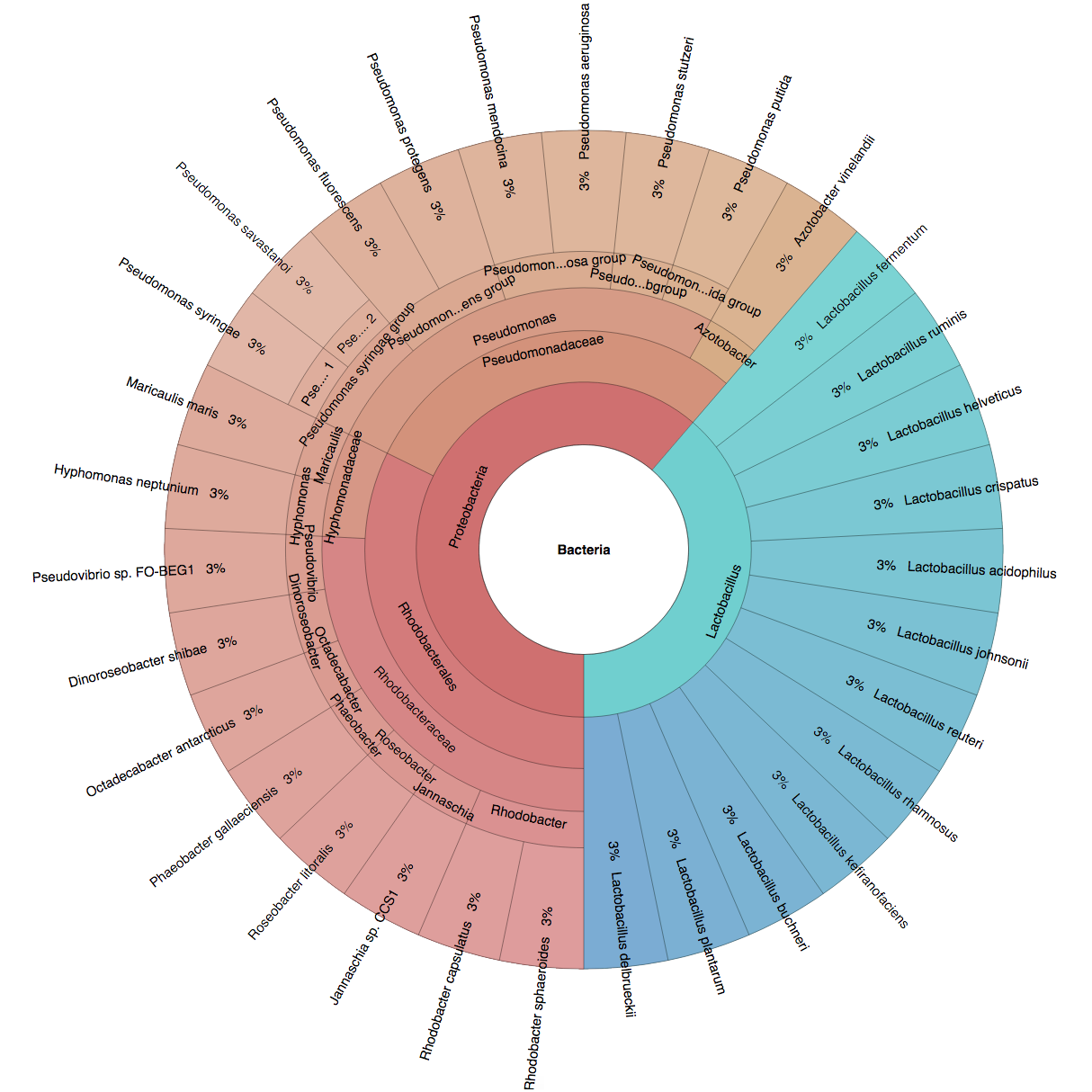
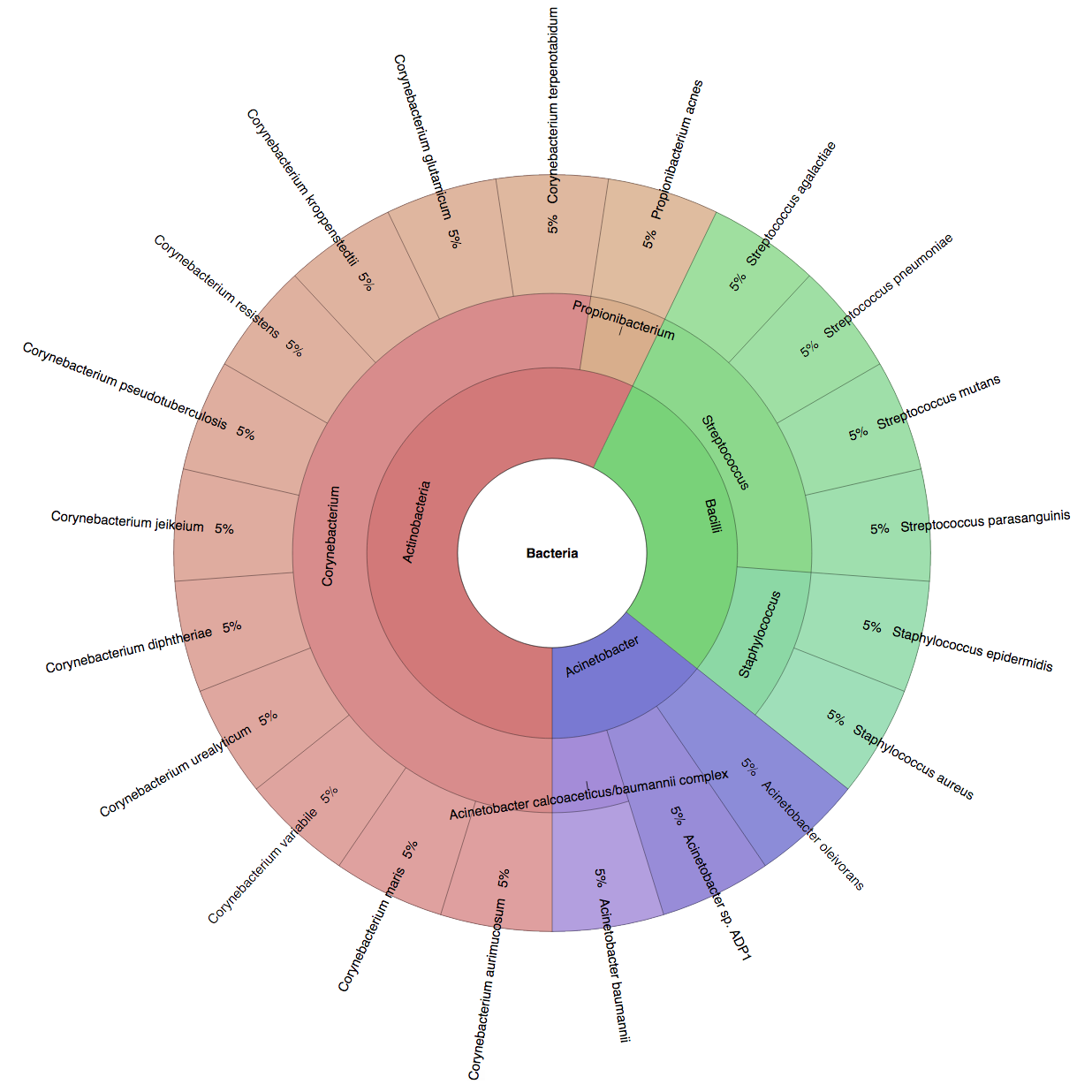
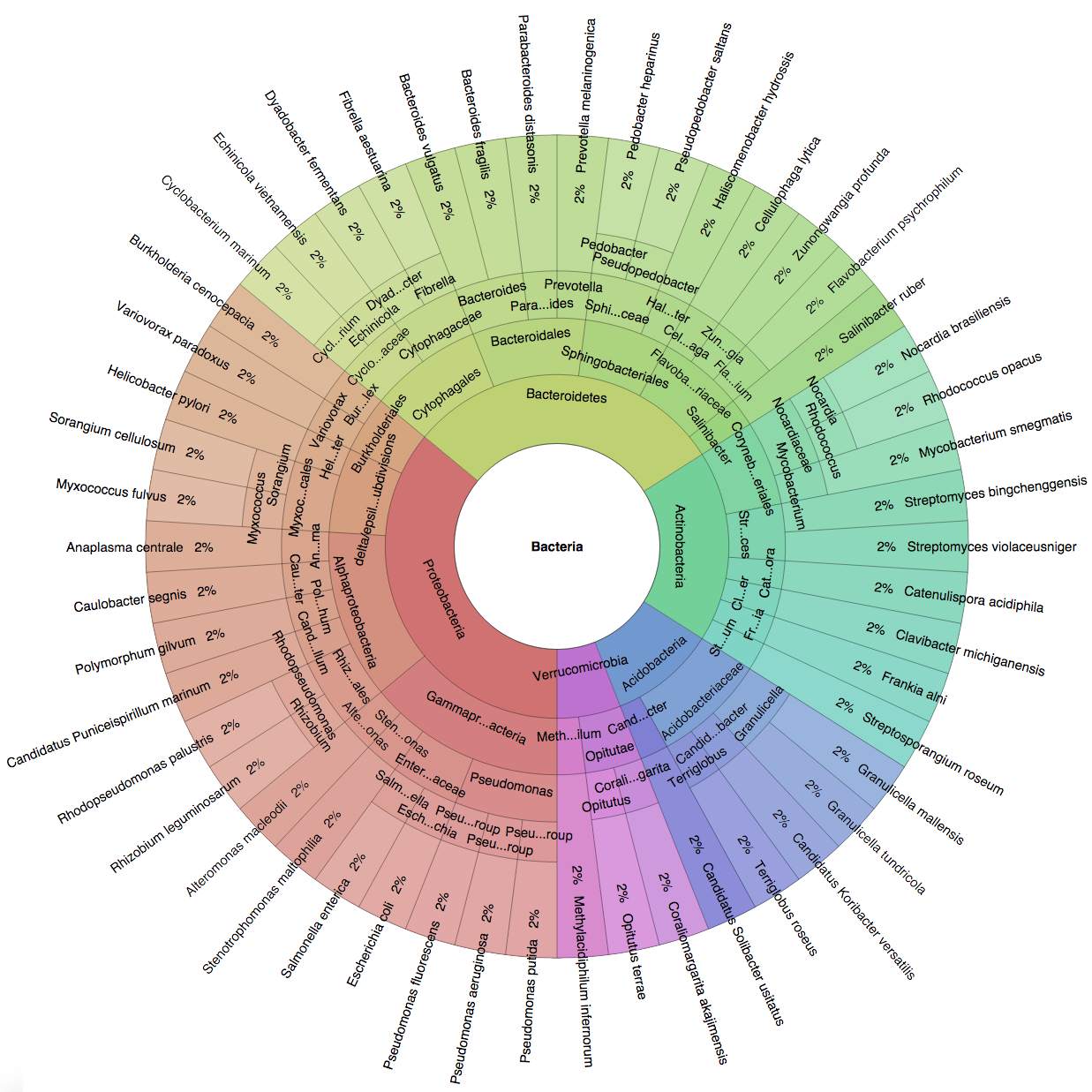
(B) In the Bioinformatics paper (2016), we used fourteen simulated datasets to evaluate the precision and sensitivity of CLARK-S.
We created six synthetic datasets (see the plots generated by Krona below for more detail) containing reads from dominant organisms found in the mouth
(Buc12), city parks/medians (CParMed48), gut (Gut20), indoor (Hou31 and Hou21) and soil (Soi50) environments.
A seventh dataset (simBA-525) containing reads randomly chosen from 525 bacterial/archaeal species was added.
These datasets are composed of short synthetic reads generated using ART
(Huang et al., 2012) with default settings.
However, observe that a short read r generated from genome gi may also exist in an other genome for the same error rate or number of mismatches.
Ignoring the possibility of ambiguity in reads classification is likely to lead to incorrect conclusions on precision and sensitivity.
In order to carry out an unbiased evaluation, we created additional datasets (called "unambiguous", see Supplemetary Note 2 of the CLARK-S paper for details),
in which no read can be mapped to more than one species with the same error rate or number of mismatches.
In total, we created fourteen datasets containing reads of size 100bp from 647 species.
| Buc12 | CParMed48 | Gut20 | Hou31 | Hou21 | Soi50 | simBA-525 |
 |
 |
 |
 |
 |
 |
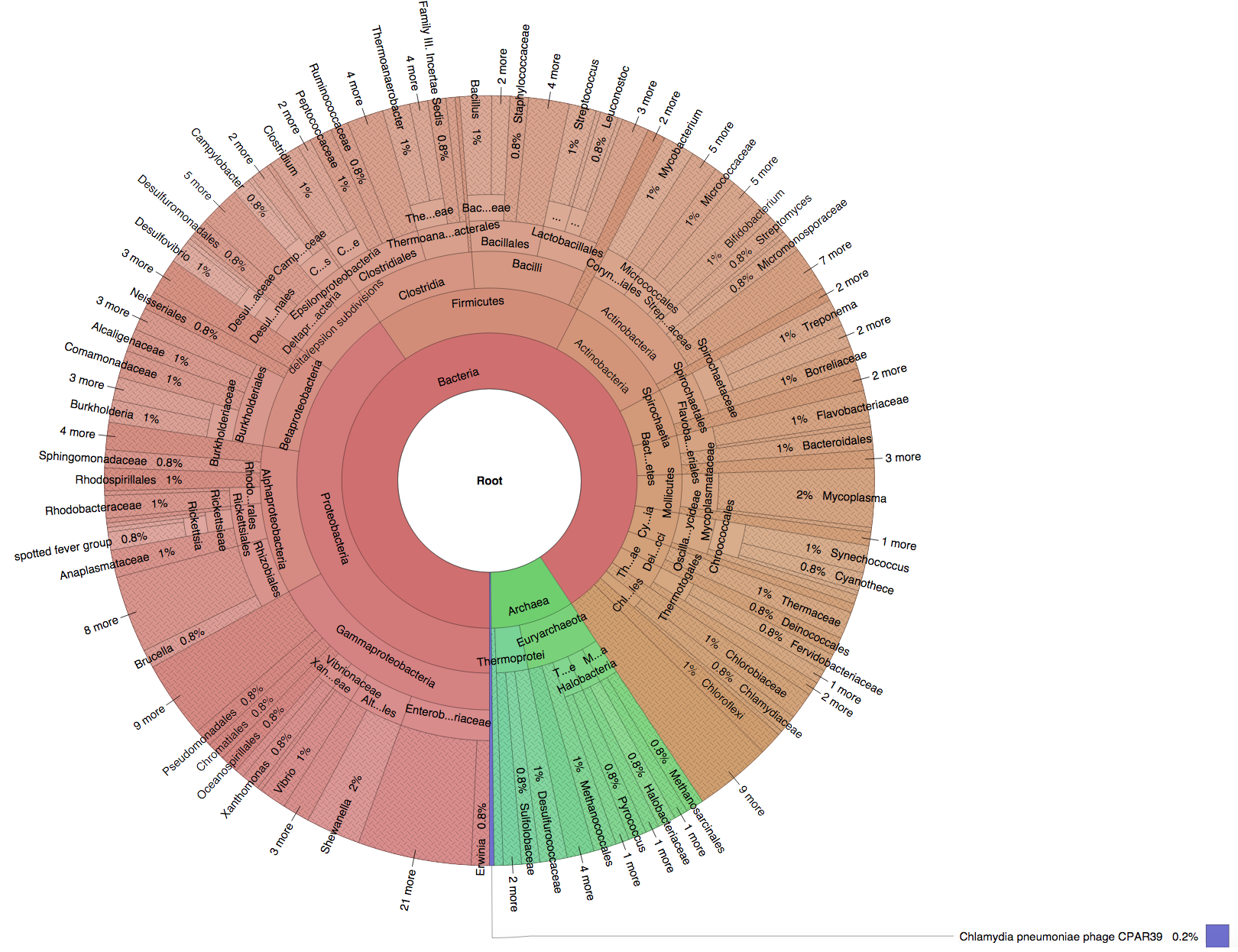 |
We also added three negative control samples containing short reads that do not exist in any genomes in the NCBI/RefSeq database. We used the precision and sensitivity metrics defined in the BMC Genomics publication to evaluate the classification performance of Kraken, CLARK and CLARK-S.
To download these datasets, go to the FAQ section (Q14).
|
ONLINE COMPUTING WITH INSIDEDNA
All three classifiers of the CLARK series are also available online in insideDNA.
This computing platform for reproducible research in bioinformatics, genomics and life science allows
you to process with a great ease-of-use your metagenomic samples with CLARK.
Because insideDNA handles completely the computational burden, running your analysis with CLARK on public data
can be carried out now simply with your laptop and an internet access.
To know more about insideDNA, see this recent article in GenomeWeb or visit the website.
|
REFERENCE AND CONTACT
CLARK is a joint project from the
Computer Science & Engineering department and the Botany & Plant Sciences department, of the University of California, Riverside.
To cite this work, please mention these references:
Do you have any questions about the study ? Please fell free to contact Stefano Lonardi, by email at stelo at cs.ucr.edu.
Are you using CLARK and want to share some feedback/comments about your experience ? Please visit the GoogleGroup of CLARK users
|
FUNDING
This project was funded in part by USDA, "Advancing the Barley Genome" (2009- 65300-05645) and by NSF, "ABI Innovation: Barcoding-Free Multiplexing: Leveraging Combinatorial Pooling for High-Throughput Sequencing"
(DBI-1062301) and "III: Algorithms and Software Tools for Epigenetics Research" (IIS-1302134).
|
ACKNOWLEDGEMENTS
We are thankful to the authors of NBC and Kraken for their useful advice on running their tools.
We thank Dr. Gail Rosen for constructive comments on the manuscript.
|
|

